TraVA is a database of gene expression profiles in Arabidopsis thaliana developmental stages, organs and parts based on RNA-seq analysis.
Profiles of 25 706 protein-coding genes (gene models are according to TAIR10 genome release) in 79 samples (complete list of samples with their detailed description can be found here) are presented.
There are two main blocks of analysis: 1) gene expression levels 2) differential gene expression
For any analysis, gene identifier should be entered in the window "Enter gene id". Acceptable identifiers are TAIR identifiers (ATXGXXXXX) or gene names. In latter case, use only abbreviated names (for example, LFY but not LEAFY, SAP
but not STERILE APETALA). If gene is known under different names, only TAIR gene name, not an alias, is acceptable (for example, AGL8 for AGAMOUS-LIKE8, known also as FRUITFULL, but CAL for CAULIFLOWER, known also as AGAMOUS-LIKE10).
For more information on gene names refer to https://www.arabidopsis.org/portals/nomenclature/guidelines.jsp
|
|

|
To start enter your gene ID in ATxGxxxxx format.
|
|
|
|
Working with gene expression levels.
This block allows you to see the expression levels of a given gene or multiple genes
in each sample, without making comparisons between samples. In case of multiple genes,
though theoretically there is no upper limit on gene number but we would not advise to search more than 10-15 genes.
|
|
|
|
Differential gene expression.
|
|

|
|
This block allows to see whether there are the statistically significant differences in expression of a given gene between samples. You can choose a software used for differential expression detection(DESeq, DESeq2 and BaySeq)
here
|
|
|
To proceed to the analysis, click on a box of the sample which be used as “control” during analysis.
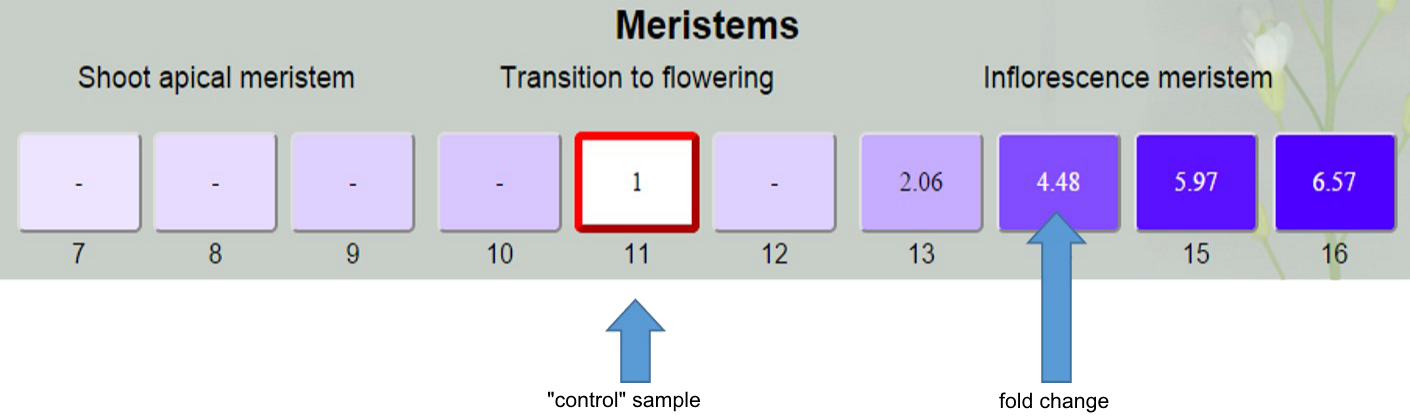
After that you will see fold changes of expression level in sample boxes. “Control” sample is indicated be the red frame and have fold change value 1. For samples that do not significantly differ from “control” fold change is “-“.
|
|
|
|
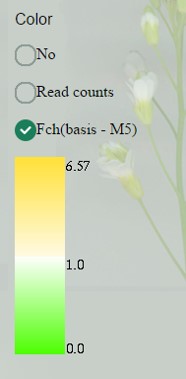
Coloring of sample boxes (which is read counts-based by default) can be changed to fold change-based. At this case, values below 1, indicating down-regulation in this sample comparing to “control”, are colored by graded green, more intense for lower values (close to 0).
|

|
|
Values above 1 (up to maximum fold change) are colored in orange, more intense for higher values.
|
|
|
Now you can go back to read counts display and still see coloring by fold change. “Control” sample is indicated as (basis - **).
|
|